
Recent CAR-T clinical studies in glioma and glioblastoma patients
Recent papers have hinted at the exciting promise of cell therapy to treat aggressive CNS malignancy, but also reveal critical issues limiting success.
A brief introduction
Glioblastoma (GBM) and high-grade glioma (HGG) are fast-growing and deadly tumors that spread within normal brain tissue and are therefore difficult to eradicate. Surgery, chemotherapy and radiation therapy are the standard treatment options, sometimes paired with anti-VEGF antibody (bevacizumab) therapy, but these regimens all fail to produce durable responses for patients. Median OS for GBM at first recurrence ranges from 5.5 to 12.6 months and post-bevacizumab is between 3 and 4 months. Thus, patients progress rapidly after therapy and experience poor quality of life prior to eventual death. The unmet need for patients with these cancers is extreme.
Chimeric antigen receptor T cells (CAR-T) are directed to attack tumor cells via recognition of tumor antigens and have been remarkably successful in the treatment of B cell cancers including leukemias, lymphomas and myeloma. In the following papers CAR-Ts were targeted to CNS tumor antigens in order to evaluate safety, preliminary efficacy and biomarkers of therapy such as CAR-T cell persistence and distribution in the patients and the induction of pro-inflammatory cytokines associated with anti-tumor immunity, such as interferon-gamma (IFNg).
Three interesting papers
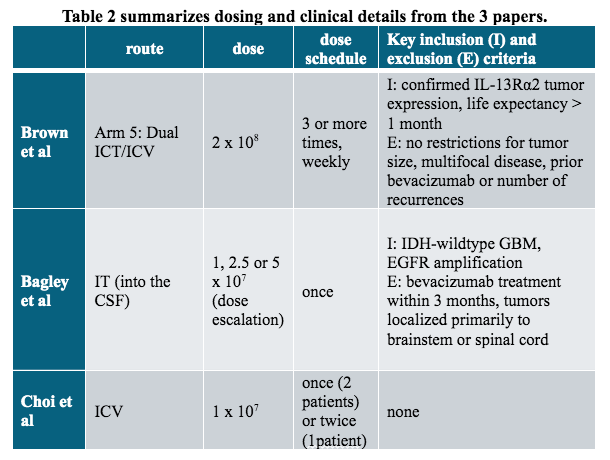
Following is a brief discussion of the papers in which unique characteristics of each CAR and T cell technology are considered in light of the reported outcomes. The investigators used a variety of tumor targeting strategies and CAR-T manufacturing techniques (Table 1) and different CAR-T delivery methods and doses (Table 2). All CAR vectors used 4-1BB and CD3z signaling domains to trigger cell activation.
1 - Choi et al (2024). Intraventricular CARv3-TEAM-E T Cells in Recurrent Glioblastoma. NEJM. DOI: 10.1056/NEJMoa2314390. (from Mass General Hospital)
Choi et al. have opened their clinical study with three recurrent GBM patients. GBM is a cancer whose cells that rely on growth factor EGFR signaling for proliferation and survival signal, indeed many GBM patients have EGFR gene amplification and/or have mutations in the EGFR sequence that support constant positive signaling.
Patients were treated with CAR-T cells engineered to recognize EGFRvIII which is an EGFR splice variant expressed by tumor cells. The CAR-T cells were also engineered to secrete a bispecific T cell engager protein (BiTE) that recognizes the normal form of EGFR (the BiTE is an anti-CD3/anti-EGFR engineered from antibody sequences). CAR-T cells were delivered to the patients in a single intraventricular infusion (ICV, see Table 2). Of note, analysis of the infused cells showed that the BiTE protein bound to the CAR-T cells, thus 93% of the CAR-T cells were preloaded with the BiTE (their Figure 4b). It follows that each CAR-T cell initially could be activated in two ways: through the CAR domain upon binding to EGFRvIII and through the preloaded BiTE upon binding to EGFR. It is unclear if the BiTE alone also activated normal CD3-positive T cells in the patients although this is theoretically possible.
Treatment with CARv3-TEAM-E T cells did not cause any severe adverse events and no dose-limiting toxicity was observed in these 3 patients. Tumor regression occurred rapidly post-CAR-T infusion and one patient had a durable response through 5 months as measured by MRI and by measurement of EGFR and EGFRvIII transcripts in the cerebrospinal fluid (CSF).
Quantification of CAR-T cells and BiTE-loaded CAR-T cells showed transient cell persistence in CSF samples with rapid decline through day 28 (see their Figure 4C). Patient 2 - who obtained the longest response - had the lowest expansion in the CSF of both CAR-T cells and of BiTE-loaded CAR-T cells (see Figure 4C in their paper) but had the highest measured expression of IFNg that is expressed by T cells, including CAR-T cells, when they are activated. Thus, presence of the CAR-Ts and the secreted BiTE in the CSF was not correlated with clinical response in this small study.
In the follow-up, this study will enroll recurrent and newly diagnosed GBM patients for a multi-dose regiment of CARs given weekly x 6 weeks (NCT05660369).

2 - Bagley et al. 2024. Intrathecal bivalent CAR T cells targeting EGFR and IL13Rα2 in recurrent glioblastoma: phase 1 trial interim results. Nat. Med. https://doi.org/10.1038/s41591-024-02893-z (University of Pennsylvania)
Bagley and colleagues treated 6 refractory GBM patients with intrathecally (IT) delivered bispecific CAR-T-cells targeting EGFR epitope 806 (specific for tumor cells) and IL13Rα2, a known GBM antigen (Table 1). The use of binders to two different antigens is to address the known problem of tumor antigen heterogeneity leading to relapse as has been seen when only IL13Rα2 was targeted. EGFR epitope 806 is known to be expressed on 50–60% of GBM patient tumor cells and IL13Rα2 is known to be expressed on 50–75% of GBM patient tumor cells. Therefore, one or both of the antigens is expected to be present on nearly all GBM tumor cells.
As in the MGH study, reductions in MRI enhancement and measured tumor size were observed in all patients at early imaging timepoints although none met the criteria for clinical response (tumor volume reduced > 30%). Tumor size was reduced on the first MRI scan obtained 24–48 h after CAR T cell administration, and partial tumor regression maintained at day 28+ in a subset of patients including one patient with stable disease beyond 4 months post-CAR treatment.
Using biomarker analyses similar to the prior study, CAR-T cells and inflammatory cytokines were detected in the CSF of all patients. Peak CAR-T cellularity in the CSF was observed between days 1 and 7 post-CAR, reaching an average of 109,235 copies of CAR per ug of genomic DNA. Note these levels are 2x-10x higher than that obtained in the MGH study. Possibly because such levels were achieved, CAR-T cells were also found in the peripheral blood in all patients.
Resistance mechanisms thought to limit anti-GBM immunity include antigen loss, as discussed above, a highly immunosuppressive tumor microenvironment (TME) and T cell dysfunction of the CAR-T cells in the infused product or within the TME. In line with the next study, the UPenn group plans to characterize CAR-T cell effector/memory phenotypes prior to infusion to look for correlations with clinical outcomes (NCT05168423).
3 - Brown et al (2024). Locoregional delivery of IL-13Rα2-targeting CAR-T cells in recurrent high-grade glioma: a phase 1 trial. Nat Med. https://doi.org/10.1038/s41591-024-02875-1 (City of Hope)
The City of Hope team enrolled 65 patients with recurrent HGG, the majority of which had recurrent GBM. 58 patients were evaluable.
The study tested 3 routes of T cell administration: intratumoral (ICT), intraventricular (ICV) and dual (ICT/ICV). Arm 5 of the study used dual ICT/ICV delivery and an optimized manufacturing process that allowed expansion to the clinical maximum feasible dose of 2 × 108 cells. Arm 5 CAR-T cells were CD62L+ enriched naive, stem cell memory and central memory T cells (Tn/mem). Evaluable patients received at least 3 weekly infusions of CAR-T cells and additional infusions were allowed until disease progression. Table 2 summarizes these details.
Stable disease or better was achieved in 50% (29/58) of patients, with two partial responses (PR: >30% reduction in tumor diameter) one complete response (CR: 100% reduction in tumor size) and a second CR after additional CAR-T cycles off protocol. Focusing on Arm 5 that used optimized CAR-T cell manufacturing and Tn/mem cells, 9/21 patients (43%) survived beyond 12 months and median OS was 10.2 months.
CAR-T cells were detected in the CSF in the most patients 1 day post infusion for at least one cycle, and for a subset of patients, ≥7 days post infusion, reaching a maximum of ~700 copies per ug of DNA in arm 5, albeit with very large error bars (see their Figure 4b). CAR-T cells were also detected in peripheral blood, reaching the highest concentration in arm 5 patients. Notably, CD3+ T cell density measured in tumor biopsy samples pre-CAR infusion correlated with median OS, suggesting a pre-existing anti-tumor immune response. Finally, this study demonstrated that the CAR-T cells delivered to the CNS can persist in the CSF and traffic to the periphery (see the Discussion, below), similar to the report from University of Pennsylvania (UPenn).
This trial will move forward using Arm 5 processes (Tn/ mem manufacturing and dual ICT/ICV delivery) in three declared studies, one with or without nivolumab (anti-PD-1 antibody) and ipilimumab (anti-CTLA4 antibody) to counter immunosuppression (NCT04003649), one for pediatric patients (NCT04510051) and one for leptomeningeal GBM, ependymoma, or medulloblastoma patients (NCT04661384).
Discussion: the papers
What we see here are signs of promise in the development of CAR-T therapy for a deadly solid tumor type. Each study presents 1 or more patients in which a number of positive signals are detected: reduction in tumor volume, expansion of CAR-T cells, induction of IFNg and other pro-inflammatory cytokines, and in the final study by Brown et al., association with CD3 T cell infiltration of the tumor itself. The challenges encountered are consistent with current views regarding antigen-loss relapse and the immunosuppressive nature of the GBM TME.
The story acquires a little more depth as we look across the studies.
In the first paper from MGH, two different anti-EGFR targeting strategies are used, one to target wildtype (ie. normal) EGFR, and one to target a splice variant EGFRvIII that is expressed only by tumor cells. Two modalities are also used, the CAR-T cell itself (anti-EGFRvIII) and the secreted BiTE, an anti-CD3/anti-EGFR protein that activates CAR-T cells by binding to CD3 and binding to the tumor cell-expressed EGFR. It will be interesting to know if the BiTE can activate CD3+ T cells within the patient in addition to activating the CAR-T cells. Dual antigen-targeting should prevent loss of CAR response due to loss of antigen, nonetheless, CAR persistence in this study was limited, as were the anti-tumor responses. Patient 1 lost EGFRvIII expression and temporarily lost EGFR expression and patients 2 and 3 eventually lost expression of both antigens. In this small study it is difficult to draw any correlations between biomarkers and responses.
The UPenn study is another dual antigen-targeting study, using a bispecific CAR to attack the EGFRVIII epitope 806 and antigen IL-13Ra2. They enrolled 6 patients and triggered robust CAR-T cell expansion that peaked at day 7 and was still measurable at day 28 in the CSF and in peripheral blood samples. Despite the robust CAR expansion and concomitant IFNg secretion, anti-tumor responses as measured by MRI were limited in extent to stable disease (SD).
Finally, the City of Hope paper reports on the largest number of patients, while using a variety of strategies. In Arm 5, the use of a selected CAR T phenotype (Tn/mem) was combined with two different means of CAR delivery, and perhaps most critically, a multiple dosing paradigm with at least 3 weekly doses given at the outset.
This paper presents several interesting biomarker observations. CAR persistence was limited after each cycle, at ~ 7 days, and not markedly higher in the CSF of patients in Arm 5 as compared to the other arms in the study. IFNg-related cytokines (signature: IFNg, CXCL9 and CXCL10) were markedly higher in the CSF of Arm 5 patients. Also Arm 5 patients showed the most robust CAR cellularity and IFNg-related signature in the peripheral blood. Finally CD3-positive cellularity in the biopsy samples of patients was correlated with responsiveness regardless of arm of therapy – there were 14 CD3-intermediate and only 3 CD3-high patients represented – but they presented the best survival probability (see their Figure 4A,B).
What can we make of these observations?
IFNg is a cytokine that is directly cytotoxic to tumor cells and plays an outsized role in anti-tumor immunity. CXCL9 and CXCL10 expression is induced by IFNg stimulation of mainly non-immune cells like fibroblasts and epithelial cells but also tumor cells. These chemotactic cytokines, or chemokines, regulate immune cell migration, differentiation, and activation. Immune reactivity occurs through recruitment of immune cells including T cells. Tumor-infiltrating T cells (whether normal T cells or engineered CAR-T cells) are critical for clinical response – this just means that T cells have to get into the tumor to initiate killing. This recruitment can be induced by CXCL9 and CXCL10 that bind to receptors expressed on the T cell surface. These same chemokines play similar roles organizing the distribution of immune cells in immune organs such as lymph nodes and spleen.
Some negative biomarker conclusions: 1) Across these 3 studies CSF CAR-T cellularity seems of limited use in predicting clinical response, although there may be useful data within Arm 5 of the City of Hope study, if it were broken out by patient. 2) CAR-T persistence in all studies is limited. 3) Multi-antigen targeting does not appear to solve the persistence issue at least in these HGG and GBM cancers.
Some positive biomarker conclusions: 1) CAR-T cell response is associated with pre-existing anti-tumor immunity as represented by normal CD3+ T cells within the tumor as measured in biopsy samples pre-CAR-T infusion. 2) CAR-T response is associated with a IFNg signature that includes the T cell-attracting chemokines CXCL9 and CXCL10. The presence of T cells within the tumor bed indicates that an ongoing, if ultimately unsuccessful, anti-tumor immune response was triggered within lymph nodes that drain the CNS through the lymphatic system. It is tempting to speculate that successful CAR-T treatment can restimulate the natural immune system by directly killing tumor cells, allowing antigen and antigen presenting cells (APCs) into the lymph nodes.
How do these findings fit into the larger cell therapy and immuno-oncology landscapes
An intriguing array of technologies are on display in these papers targeting these intractable and deadly solid tumors. It is tempting to conclude that deep and durable responses may soon be achieved for these patients, but before this can happen the CAR-T persistence issue needs to be solved. Looking ahead, the confluence of diverse technologies will have to continue, and with this in mind I’d look to several areas to contribute. One of these is the CAR vaccine technology space as represented by BioNTech, Elicio and many others. These technologies seek to place CAR-T antigens within the immune system itself, by targeting lymph nodes either with mRNA-containing nanoparticles (BioNTech) or with the CAR-targeting antigen bound to serum proteins that are directed to lymph nodes, allowing APC in lymph nodes to display the antigen directly to CAR-T cells. As proof-of-concept, BioNTech has shown increases in CAR-T numbers in patients upon receipt of their “CAR-VAX”. Elicio has reported promising Phase 1 results in pancreatic cancer.
Another area poised to deliver benefits are the ‘armored’ CARs that can produce additional cytokines or chemokines to broaden the immune attack. Cytokines such as IL-12 and IL-18 can engage the normal immune system, essentially “waking it up” so it can contribute to anti-tumor immunity. While the cytokine delivery systems have yet to show clinical benefit, it is an area of active development and one to watch. Key to success here will be safe delivery of cytokines that are otherwise toxic if given systemically. We point to the deal just announced between Xilio and Gilead as an example of the importance of safe delivery technology.
Finally, there are technologies that directly leverage bona fide immune cell interactions with CAR-T cells to improve CAR-T cell fitness and persistence. Seattle Children’s Hospital has reported preliminary results from the STRIVE-01 and STRIVE-02 trials in which single-antigen targeting CAR-T cells are compared to dual-antigen targeting CAR-T cells where the second antigen is CD19, an immunologically relevant antigen that is not expressed on solid tumor cells. STRIVE-01 was updated at ASCO 2023 to demonstrate that a dual-antigen CAR-T for pediatric solid tumors that recognized both B7H3, a cancer antigen, and CD19, a B cell antigen, expanded and persisted longer than a CAR-T to B7H3 only (https://doi.org/10.1200/JCO.2023.41.16_suppl.1004). This is because host B cell CD19 expression drives robust engraftment and persistence. We have shown further that engagement of a CAR-T cell with a B cell also provides the T cell with immunologically productive and supportive signals that foster T cell fitness (DOI: 10.1371/journal.pone.0247701). In the Seattle Children’s study, dual-CAR-T cells expanded 160-fold more than single-target B7H3 CAR T cells. The dual-CARs persisted for up to 8 months. Although only two SD responses were seen in 11 patients, the data point to the value of immune cell engagement to drive expansion and persistence. Gracell’s BCMA/CD19 dual CAR-T GC012F is another relevant example where CD19 targeting is being used to drive persistence in BCMA-positive multiple myeloma (MM cells do not express CD19).
These observations underscore the value of the CAR-T Engager (CTE) technology that has been developed by Aleta Biotherapeutics (www.aletabio.com) specifically to address the issues of antigen heterogeneity, relapse due to downregulation of antigen expression by tumor cells and the poor persistence and fitness of solid-tumor targeting CAR-T cells. Aleta’s solid tumor CTE features dual antigen targeting and a domain to counter immunosuppression while leveraging the positive properties of a CD19-targeting CAR-T cell. The solution provides potent cytotoxicity against solid tumor cells with CAR-T cells that are also highly fit and persistent. Aleta has built dual-antigen targeting CTEs from various solid tumor antigens including Her2, IL-13Ra2, B7H6, EGFR and B7H3. Such CTEs can be developed as lentiviral constructs for expression from CAR-T cells or as biologics for injection alongside the CAR-T cells. All dual-antigen targeting CTEs have also been built in the T cell engager format using an anti-CD3 domain, and retain high potency. If interested in working with this technology check out www.aletabio.com or email paul.rennert@aletabio.com.
SugarCone Biotech – SCB - provides clients in biotech, pharma and venture capital with strategic insights in the fields of cell therapy, gene therapy and the broader IO, oncology, immunology, autoimmune and fibrosis landscapes. To connect with SCB please reach out to rennertp@gmail.com.
This blog post is an extended version of content that was originally posted at www.aletabio.com.